OBJECTIVES
By the conclusion of this laboratory investigation, you should be able to:
- Define and properly use synthetic biology terms: chassis, system, device, redundancy.
- Define and properly use molecular genetics terms: PCR, gene expression, codon shuffling, complementation.
- Explain the role of redundancy in engineering.
- Explain how microbes can be used to make useful chemicals to meet human needs.
- Culture yeast using proper microbiology methods.
- Measure the genetic variations in a yeast population:
- Define and properly use synthetic biology terms:
- Define and properly use molecular genetics terms:
INTRODUCTION

Nature makes it look easy! Using just a few kinds of atoms, nature builds chemicals that can do all sorts of things. Even within the small collection of BioBuilder activities, you can find biologically built molecules that smell like bananas, that cut other molecules in two and that turn bacterial cells purple or green. In the experiment described here, nature’s talents as a chemist are applied to making beta-carotene, which is processed in the body into Vitamin A. Many kinds of plants and fungi make beta-carotene naturally, but animals can’t make their own from scratch. In countries where access to fresh vegetables is limited, Vitamin A deficiency is common, and in severe cases it can lead to blindness. The World Health Organization estimated that over 200 million school age children suffer from Vitamin A deficiency.
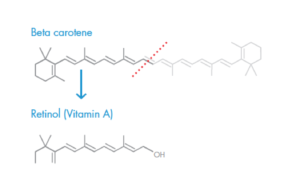
Chemical structures of beta-carotene and vitamin A
The Science and Engineering of Golden Bread
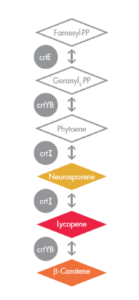
A pathway for synthesizing beta-carotene
The 2011 iGEM team from Johns Hopkins University engineered Saccharomyces cerevisiae to produce beta-carotene. This species of yeast is commonly used as a model organism in the lab as well as in bread making and brewing. The iGEM team hoped the modified yeast could be used to make nutrient-enriched loaves of “golden bread.”
Scientists had engineered Saccharomyces cerevisiae to express three genes (crtE, crtYB, and crtI) from a naturally red fungi, Xyanthophylomyces dendrorhous (the reference is linked here). The genes encode enzymes that convert a naturally produced chemical, farnesyl diphosphate, into beta-carotene. The metabolic pathway is shown in the figure on the left. The first three compounds in this pathway are colorless, and the last three are colored yellow, red, and orange, respectively. The enzymes that catalyze the reactions are shown as grey circles. Interestingly, the first reaction can be catalyzed by the enzyme crtE from the red fungus, X. dendrorhous or by the enzyme BTS1 (not shown) that is made naturally in baker’s yeast. The other two enzymes, crtYB and crtI, have no homologs in baker’s yeast and so had to be imported from X. dendrorhous, to carry out the rest of the steps in the biosynthesis of beta-carotene.
Some of the modified yeast were bright orange — a good indication that they were making beta-carotene. However, while the strain produced orange colored colonies most of the time, white, yellow and red colored colonies were also produced.
If you’d like to try to understand what’s causing the instability, jump to the “Science of Golden Bread” section.
If you’d like to try to fix this instability and engineer a strain that can make beta-carotene more reliably, then jump to the “Engineering of Golden Bread” section.
If you’d like to consider the societal implications of food engineering, then jump to the “Food for Thought” section.
Idea for modified baker’s yeast
