OBJECTIVES
By the conclusion of this laboratory investigation, you should be able to:
- Define and properly use synthetic biology terms: chassis, minimal cell, synthetic cell, sensor, color generator.
- Define and properly use molecular genetics terms: operon, gene expression, bacterial transformation.
- Explain the role of the cellular chassis in synthetic biology and engineering.
- Conduct and interpret the results of a bacterial transformation.
- Measure the growth of a bacterial population:
- Bacterial Growth Curves
- Define and properly use synthetic biology terms: Part, Open Reading Frame, iGEM
INTRODUCTION
Some Biodesign Principles
Synthetic biologists often use lab strains of E. coli because they are well understood, easy to grow and generally safe to work with, but it’s important to realize that using E. coli as a host cell for our genetic programs is a choice. This BioBuilder activity is a reminder that, just as you carefully your design for a genetic program, you also need to carefully choose the host cell, or “chassis,” that will run it. Cars are a useful analogy. Cars represent a highly engineered system of interconnected parts. Many car parts perform similar functions in different chassis, but they must be tailored to the size and function of the car they’ll run in. For instance, we might be able to move a radio from a Ford truck into a Porsche 911, but we probably can’t move the drive train or the engine.
So how are we able to move genes from one cell type to another? One trick that researchers use is to focus on strains they are familiar with, like E. coli and other domesticated cells, as chassis for experiments. In the case of E. coli, most of the strains that are used in research labs are one of two kinds. One strain is known as K-12 and the other B. Both strains are known to be safe and have been effectively used for genetic experiments for almost 100 years. The genetic differences between these strains seem to be minor. You can read more about the interesting history of these two strains.
But even small differences in these strains can trip up researchers, who often find that a genetic program works great in one chassis but will run differently in another. A key goal of synthetic biology is the reliable programming of cells, so fixing this lack of interoperability is an active area of current research. One approach to managing the complexity of the cellular chassis is to build a “minimal cell.” With this approach researchers are trying to whittle away at the existing genomes of our favorite lab organisms, leaving only the genes that the cell absolutely needs to survive. This allows researchers to precisely define the media needed to grow the minimal cell, reduces the chance of an emergent property (something unexpected that happens when you put two or more things together) and simplifies the modeling of its metabolism. Another approach to engineering a cellular chassis is to build a “synthetic cell.” This term has been used in a few ways by synthetic biologists. Sometimes a “synthetic cell” describes the replacing of one entire genome with the genome of another cell that’s been encoded by a DNA synthesizer. Sometimes a “synthetic cell” describes a “protocell” – that is a chemical mixture that do many of the things a cell can, like replicate DNA, divide etc.
About your experiment
One potential use of engineered bacteria is as indicator of toxic substances, such as arsenic or lead. Bacteria are able to grow themselves and can be very sensitive to the toxin levels. The 2009 Cambridge iGEM team wanted to design a bacterial toxin indicator that produced colors depending on the toxin detected. The team designed several color generator devices that could be linked to toxin sensors, and they called their engineered cells “E chromi.” One pigment they used is Violacein, a pigment produced by a handful of genes originally found in a different bacterial strain, Chromobacterium violacein. These genes were re-engineered and combined to produce either purple or green colors in E. coli.
To make the purple color, the team transferred to E. coli the entire violacein operon, which encodes five enzymes to metabolize L-tyrosine into a purple pigment. To vary the color, the team removed of the third gene in the operon sequence will cause the cell to metabolize the L-tyrosine into a green pigment. These pigments are easily visible to the naked eye so could be used to make a biosensor that turns color in response to toxins.
So now imagine that a group of engineers is manufacturing an arsenic sensor in E. coli. This group would like the intensity of purple color to vary as a function of arsenic level. Now imagine that a second group of engineers are also doing this but they use a different strain of E. coli. How sure can we be that the pigment will be expressed the same in a different chassis? Thinking back to our analogy with car chassis: would an engineer put a V-8 engine from a Lexus into a Mercedes chassis? Would the engine behave the same? Would the car?
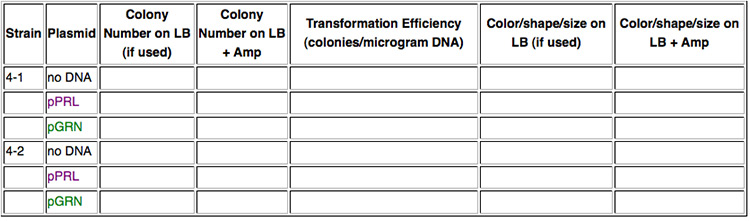
In this lab you will transform bacteria from two different strains of E. coli, in other words, two different chassis. Strain 4-1 is a K-12 strain, while strain 4-2 is a B-type strain. Into each strain you will insert plasmids containing violacein-pigment devices. One plasmid, pPRL, has the purple version of this device while the other plasmid, pGRN, has the dark green version. Otherwise, the plasmids are the same. Can we expect the devices to behave the same in each strain or will the chassis have an effect on the intensity of color produced?